

")
Recognizing Signs, Diagnosis, and Screening Protocols
 Early recognition of thalassemia symptoms is essential for timely intervention, effective disease management, and improved long-term outcomes in Thalassemia care. Patients often present with a combination of mild to severe clinical signs depending on disease type and severity.
Early recognition of thalassemia symptoms is essential for timely intervention, effective disease management, and improved long-term outcomes in Thalassemia care. Patients often present with a combination of mild to severe clinical signs depending on disease type and severity.
Common Clinical Symptoms in Thalassemia care
- Pale or yellowish (jaundiced) skin
- Persistent fatigue and low energy
- Irritability and poor concentration
- Delayed growth and development in children
- Shortness of breath during mild activity
Physical Examination Findings
- Enlarged spleen (splenomegaly)
- Liver enlargement in advanced cases
- Bone deformities in face and skull (in severe thalassemia)
- Poor weight gain in pediatric patients
Diagnostic Tests in Thalassemia care (Overview Table)
| Test | Purpose | Key Findings | Importance |
|---|---|---|---|
| Complete Blood Count (CBC) | Initial screening | Low Hb, microcytosis, hypochromia | High |
| Peripheral Blood Smear | Cell shape analysis | Target cells, abnormal RBCs | High |
| Hemoglobin Electrophoresis | Hemoglobin profiling | HbA, HbA2, HbF levels | Critical |
| Genetic Testing | Mutation identification | Alpha or beta gene defects | Definitive |
| Iron Studies | Rule out iron deficiency | Ferritin, serum iron levels | Supportive |
| MRI (Liver/Heart) | Iron overload assessment | Tissue iron deposition | Monitoring |
Role of Laboratory Testing in Thalassemia care
Laboratory investigations are the backbone of Thalassemia care, helping clinicians confirm diagnosis and plan treatment effectively.
Key Diagnostic Insights:
- CBC is the first-line screening tool
- Low hemoglobin with microcytosis strongly suggests thalassemia
- Hemoglobin electrophoresis differentiates between trait and major forms
- Elevated HbA2 and HbF indicate beta thalassemia variants
- Genetic testing provides definitive confirmation
Genetic Testing and Prenatal Diagnosis in Thalassemia care
 Genetic testing plays a critical role in confirming mutations in alpha or beta globin genes and guiding long-term management in Thalassemia care.
Genetic testing plays a critical role in confirming mutations in alpha or beta globin genes and guiding long-term management in Thalassemia care.
Key Benefits:
- Identifies exact mutation type
- Supports personalized treatment planning
- Assists in family counseling and risk assessment
- Enables early intervention strategies
Prenatal Screening Options:
- Chorionic Villus Sampling (CVS)
- Amniocentesis
These tests help detect severe forms of thalassemia before birth, allowing families to prepare for specialized care.
Monitoring and Long-Term Follow-up in Thalassemia care
Regular monitoring is a cornerstone of effective Thalassemia care, especially for patients receiving lifelong transfusions.
Essential Monitoring Components:
- Serum ferritin testing every 1–3 months
- Liver iron concentration (MRI) for iron overload tracking
- Cardiac MRI (T2)* to assess heart iron deposition
- Liver function tests to evaluate organ health
- Growth and endocrine monitoring in children
Why Regular Monitoring Matters in Thalassemia care
- Detects iron overload before organ damage occurs
- Prevents cardiomyopathy and liver complications
- Helps adjust chelation therapy effectively
- Improves survival and long-term quality of life
- Ensures safe continuation of transfusion therapy
Effective Treatment Strategies for Thalassemia care
Modern Thalassemia care focuses on a combination of lifelong supportive treatments and curative options aimed at maintaining hemoglobin levels, preventing complications, and improving quality of life. Treatment is highly individualized based on disease severity, age, and overall health condition.
Transfusion Therapy in Thalassemia care
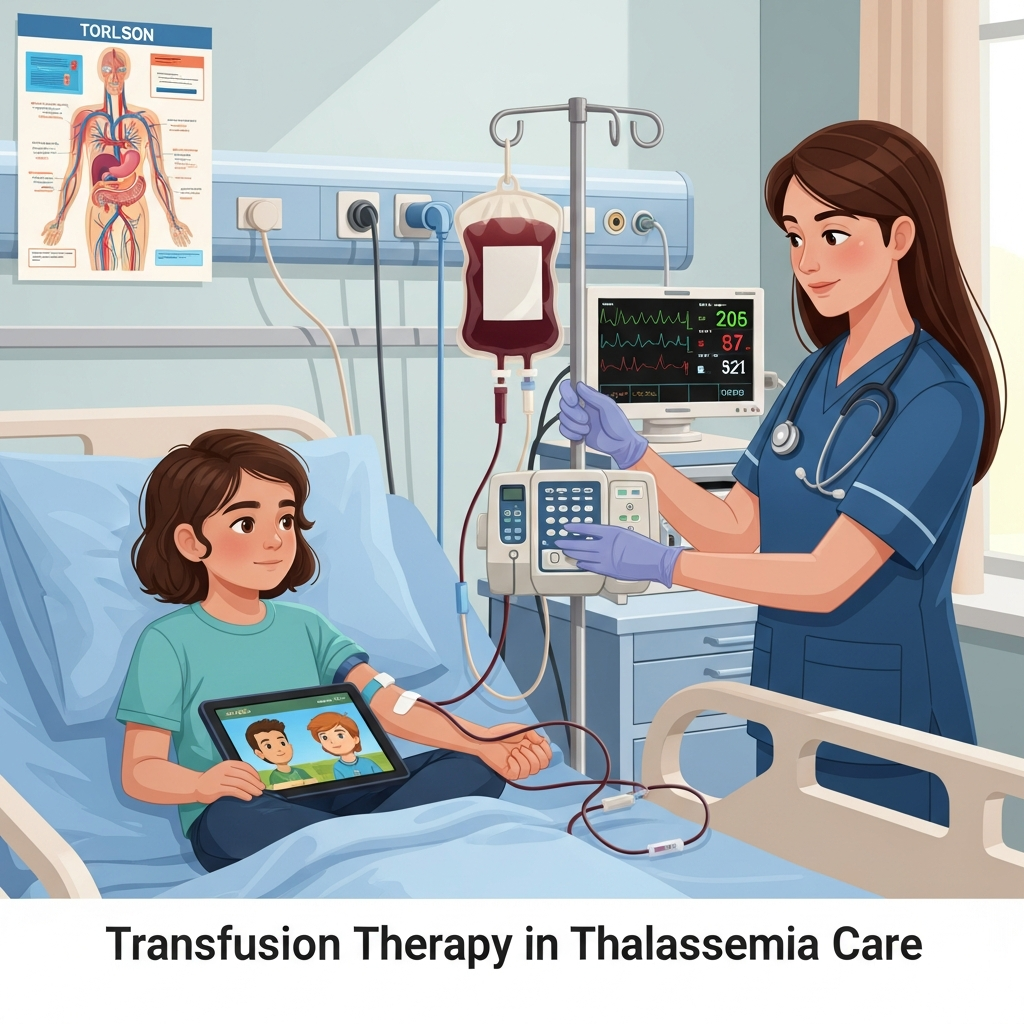 For patients with beta thalassemia major or severe forms of alpha thalassemia, regular red blood cell transfusions remain the foundation of effective Thalassemia care.
For patients with beta thalassemia major or severe forms of alpha thalassemia, regular red blood cell transfusions remain the foundation of effective Thalassemia care.
Key Clinical Goals:
- Maintain pre-transfusion hemoglobin levels between 9–10.5 g/dL
- Reduce chronic fatigue and weakness
- Support normal physical and cognitive development in children
- Prevent skeletal deformities and bone marrow expansion
- Improve overall oxygen delivery to tissues
Standard Transfusion Schedule:
- Typically every 2 to 4 weeks
- Based on individual hemoglobin response
- Requires strict blood matching and screening
Clinical Benefits:
- Improved energy levels and daily functioning
- Reduced organ stress and complications
- Better growth outcomes in pediatric patients
- Enhanced long-term survival rates
Iron Chelation Therapy in Thalassemia care
While transfusions are life-saving, they also lead to iron accumulation, making iron chelation a critical component of Thalassemia care.
Why Iron Chelation is Needed:
- Prevents iron overload in:
- Liver
- Heart
- Endocrine glands
- Reduces risk of fibrosis and organ failure
- Protects long-term organ function
Common Iron Chelators:
| Drug | Administration | Key Feature |
|---|---|---|
| Deferoxamine | Subcutaneous infusion (8–12 hours/night) | Highly effective but intensive |
| Deferasirox | Oral once daily | Convenient and widely used |
| Deferiprone | Oral multiple doses daily | Strong cardiac iron removal |
Monitoring in Thalassemia care:
- Serum ferritin levels (regular testing)
- MRI-based liver iron concentration
- Cardiac MRI (T2*) for heart iron
- Dose adjustments based on iron burden
Key Benefits:
- Prevents cardiomyopathy
- Protects liver from long-term damage
- Maintains endocrine function
- Improves overall survival outcomes
Hematopoietic Stem Cell Transplantation in Thalassemia care
Hematopoietic stem cell transplantation (HSCT) is currently the only established curative option in Thalassemia care, particularly for thalassemia major.
Who is Eligible:
- Young patients with severe disease
- Individuals with an HLA-matched sibling donor
- Patients without significant organ damage
Success Outcomes:
- Survival rates exceed 80% in early childhood transplantation
- Best outcomes achieved when performed at younger ages
Potential Risks:
- Graft-versus-host disease (GVHD)
- Severe infections due to immune suppression
- Transplant-related complications
- Long recovery and intensive follow-up required
Post-Transplant Care:
- Continuous monitoring of immune recovery
- Infection prevention strategies
- Long-term organ function assessment
- Regular hematology follow-up
Key Insight in Thalassemia care
Effective treatment in Thalassemia care is not limited to a single approach. Instead, it combines:
- Lifelong transfusion support
- Careful iron overload management
- Curative stem cell transplantation (when possible)
- Continuous monitoring and multidisciplinary follow-up
Optimizing Lifestyle and Nutrition for Thalassemia Care
 Alongside medical treatment, Thalassemia care strongly depends on lifestyle choices, nutrition, and emotional well-being. A holistic approach helps patients maintain energy levels, protect organ health, and improve long-term quality of life.
Alongside medical treatment, Thalassemia care strongly depends on lifestyle choices, nutrition, and emotional well-being. A holistic approach helps patients maintain energy levels, protect organ health, and improve long-term quality of life.
Nutrition and Diet in Thalassemia care
A well-balanced diet supports red blood cell production, strengthens immunity, and helps maintain overall body function in Thalassemia care.
Key Dietary Principles:
- Focus on high-quality protein intake
- Ensure adequate vitamins and minerals
- Maintain balanced energy levels based on age and activity
- Avoid unnecessary iron overload from diet and supplements
Recommended Foods:
Protein-rich sources:
- Lean chicken and poultry
- Fish and seafood
- Eggs
- Lentils, beans, and legumes
Vitamin and mineral support:
- Leafy green vegetables (folate-rich)
- Nuts and seeds
- Whole grains
- Fruits rich in antioxidants
Bone health support:
- Milk and dairy products
- Fortified plant-based milk
- Calcium and vitamin D sources
Important Dietary Precautions in Thalassemia care
- Avoid iron supplements unless prescribed
- Limit excessive red meat consumption
- Avoid iron-fortified products without medical advice
- Do not self-medicate with vitamins
- Always follow a diet plan guided by a healthcare professional
Lifestyle Management in Thalassemia care
Healthy lifestyle habits are essential to support circulation, energy, and overall physical stability in Thalassemia care.
Recommended Physical Activities:
- Walking at a comfortable pace
- Swimming (low-impact exercise)
- Yoga and stretching routines
- Light cycling depending on energy levels
Exercise Guidelines:
- Avoid overexertion during fatigue episodes
- Adjust activity based on hemoglobin levels
- Maintain consistency rather than intensity
- Always seek medical clearance before intense exercise
Additional Lifestyle Practices:
- Maintain proper hydration
- Practice sun protection to reduce skin-related risks
- Ensure good dental hygiene to prevent infections
- Follow regular sleep schedules for energy balance
Psychosocial Support in Thalassemia care
Emotional and mental health support is a vital part of comprehensive Thalassemia care, as chronic illness can significantly affect psychological well-being.
Common Challenges:
- Anxiety related to lifelong treatment
- Depression due to fatigue and health limitations
- Social stigma or misunderstanding
- Stress from frequent hospital visits
Coping Strategies in Thalassemia care
- Psychological counseling and therapy
- Peer support groups (online and offline)
- Mindfulness, meditation, and breathing exercises
- Participation in hobbies and social activities
- Family involvement in care routines
Education and Awareness in Thalassemia care

Ongoing patient education improves adherence and reduces complications.
Key Focus Areas:
- Importance of regular transfusions and follow-ups
- Early warning signs of complications
- Medication adherence (especially iron chelation)
- Emergency care awareness
Modern Support Tools:
- Mobile health applications
- Telemedicine platforms
- Patient monitoring portals
- Digital reminders for treatment schedules
Social and Financial Support in Thalassemia care
Support systems play a major role in improving accessibility and long-term outcomes.
- Assistance from social workers and case managers
- Help with treatment cost and insurance processes
- Transportation support for hospital visits
- School and workplace accommodation plans
Thalassemia care exemplifies the power of a comprehensive, patient-centered approach. From precise genetic diagnostics and tailored transfusion schedules to rigorous iron chelation protocols, every aspect of treatment is designed to optimize health outcomes. Equally important are lifestyle adaptations—balanced nutrition, appropriate exercise, and emotional support—that sustain daily well-being. By partnering with a multidisciplinary care team, leveraging authoritative resources, and engaging in proactive education, patients and families can navigate the challenges of thalassemia with confidence. This holistic framework not only addresses medical needs but also nurtures psychological resilience and social integration, ensuring that those affected by thalassemia can thrive and achieve their fullest potential in today’s complex healthcare environment.







{kind=link}