


Alpha thalassemia is a genetic blood disorder affecting hemoglobin production, causing mild to severe anemia. Early diagnosis and proper treatment help manage symptoms, while ongoing gene therapy research offers hope for more effective and potential future cures.
Blood is the lifeline of the human body, carrying oxygen to every tissue and organ to keep them functioning properly. At the core of this delivery system is hemoglobin, a complex protein found inside red blood cells. When genetic mutations disrupt the production of this vital protein, it can lead to a group of blood disorders known as thalassemia.
One of the primary forms of this condition is alpha thalassemia. Because the severity of this disorder depends entirely on your genetic makeup, people experience it in vastly different ways. Some individuals carry the genes without ever feeling sick, while others face severe, life-threatening complications that require immediate and lifelong medical care.
Recognizing alpha thalassemia symptoms early on is crucial for getting the right medical support. If you or a loved one have a family history of blood disorders, understanding the signs and knowing what to expect can make a massive difference in your quality of life. This comprehensive guide will walk you through the genetic causes, the wide range of symptoms, the diagnostic process, and the most effective treatment options available today.
Introduction to Alpha Thalassemia
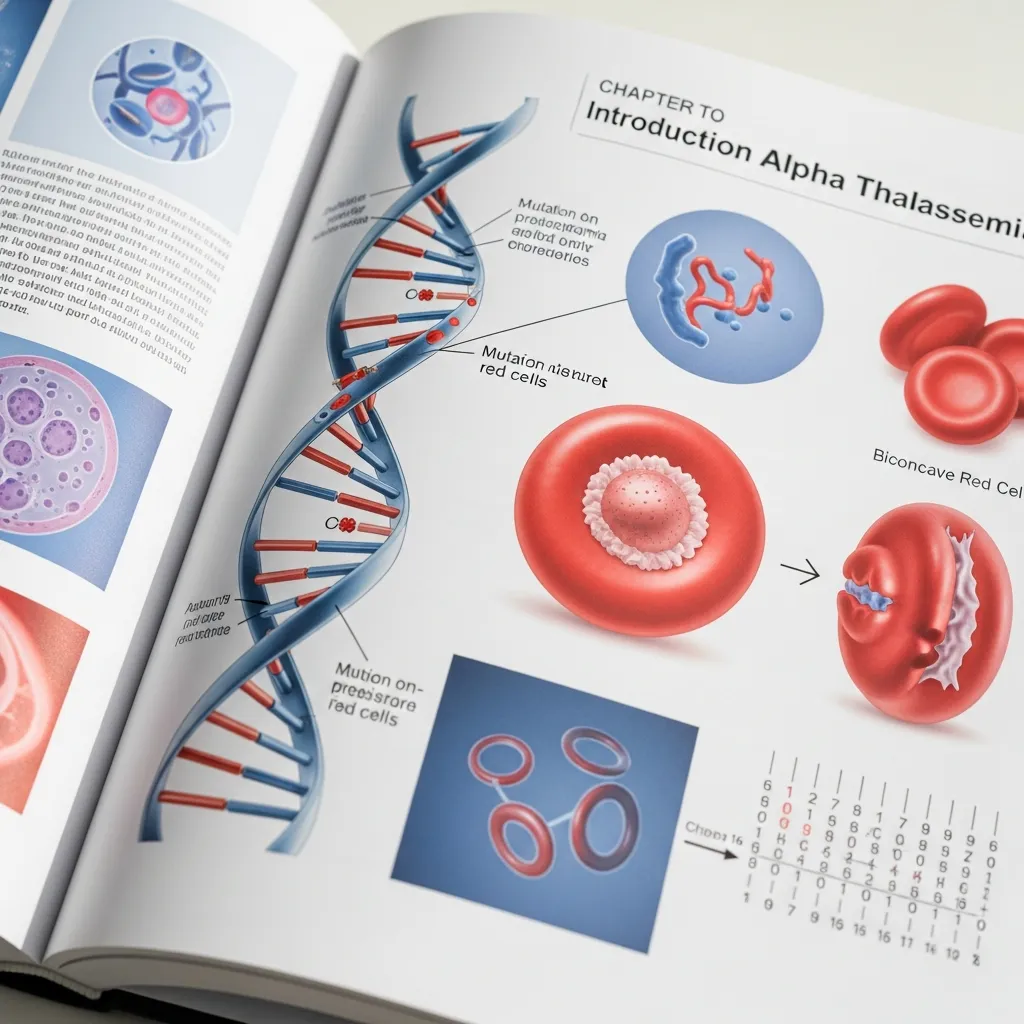 To truly understand how this condition affects the body, it helps to start with the basics of how our blood works and where the problem originates.
To truly understand how this condition affects the body, it helps to start with the basics of how our blood works and where the problem originates.
What is Alpha Thalassemia?
Alpha thalassemia is an inherited blood disorder that reduces the body’s ability to produce alpha-globin. Hemoglobin is made up of different building blocks, primarily alpha-globin and beta-globin chains. When your body cannot produce enough alpha-globin, the remaining beta-globin chains clump together. These abnormal clumps damage red blood cells, causing them to break down faster than normal. This rapid destruction leads to anemia, a state where your body simply does not have enough healthy red blood cells to transport oxygen effectively.
Genetic basis and inheritance patterns
Your genes act as the instruction manual for building proteins. The instructions for making alpha-globin are located on chromosome 16. Every person inherits four of these alpha-globin genes—two from their mother and two from their father. Alpha thalassemia occurs when one or more of these four genes are missing or mutated. The condition is passed down from parents to children. If both parents are carriers of defective genes, their children have a much higher risk of inheriting a more severe form of the disorder.
Prevalence and global impact
While this condition can affect anyone, it is most frequently found in specific parts of the world. Alpha thalassemia is particularly common among populations in Southeast Asia, Southern China, the Middle East, the Mediterranean region, and parts of Africa. Global health organizations estimate that millions of people worldwide carry at least one mutated alpha-globin gene. Because global migration has spread these genetic traits, screening and awareness are now vital public health priorities everywhere.
Understanding Alpha Thalassemia Symptoms
The missing number of genes directly dictates how the body responds. Because of this, alpha thalassemia symptoms range from completely unnoticeable to exceptionally severe.
Mild Alpha Thalassemia (Alpha Thalassemia Minima/Trait)
When a person is missing one or two alpha-globin genes, they have a mild form of the condition.
- Often asymptomatic or very mild anemia: Most people with one missing gene (silent carriers) feel entirely healthy. Those with two missing genes (alpha thalassemia trait) might experience very mild fatigue or slight paleness, but they generally live normal, active lives.
- Detection during routine blood tests: Because these individuals rarely feel sick, the condition is usually discovered by accident. A doctor might notice unusually small red blood cells during a routine annual blood test and order further screening to confirm the trait.
Hemoglobin H Disease (HbH Disease)
If a person inherits three missing or defective genes, they develop Hemoglobin H (HbH) disease. This leaves them with only one functioning alpha-globin gene, leading to a noticeable shortage of healthy hemoglobin.
- Moderate to severe anemia: Patients frequently experience chronic fatigue, weakness, and shortness of breath during physical activity.
- Splenomegaly (enlarged spleen): The spleen works overtime to filter out the damaged red blood cells. This extra workload causes the spleen to swell, which can lead to abdominal pain or a feeling of fullness.
- Jaundice and gallstones: As red blood cells break down rapidly, they release a yellow pigment called bilirubin. This causes the skin and the whites of the eyes to turn yellow (jaundice) and significantly increases the risk of developing gallstones.
- Bone changes (facial and skull): To compensate for the lack of red blood cells, the bone marrow expands to produce more. This expansion can cause the bones in the face and skull to widen or become brittle.
- Growth retardation: Children with HbH disease often experience slower physical growth and delayed puberty due to the body’s chronic lack of oxygen.
Hydrops Fetalis (Barts Hydrops)
The most severe form of the disorder occurs when all four alpha-globin genes are missing.
- Severe form, often fatal before or shortly after birth: Without any alpha-globin, normal hemoglobin cannot form. The fetus develops a type of hemoglobin called Hemoglobin Barts, which holds onto oxygen so tightly that it cannot deliver it to the body’s tissues.
- Extreme anemia, widespread edema, and organ failure: The fetus suffers from severe oxygen deprivation, leading to massive fluid buildup (edema), heart failure, and severe organ damage in thalassemia.
- Maternal complications: This condition also poses severe health risks to the pregnant mother, including high blood pressure, premature delivery, and dangerous bleeding during birth.
The Spectrum of Severity: From Silent Carriers to Life-Threatening Conditions
 Understanding where a patient falls on the severity spectrum helps doctors create the best possible care plan.
Understanding where a patient falls on the severity spectrum helps doctors create the best possible care plan.
Silent Carrier State
With only one missing gene, silent carriers show no physical alpha thalassemia symptoms. Their red blood cells might be slightly smaller than average, but they do not have anemia. Identifying silent carriers is mostly important for family planning, as they can pass the missing gene to their children.
Alpha Thalassemia Trait
People missing two genes have the alpha thalassemia trait. They might have mild anemia, which is sometimes misdiagnosed as iron deficiency. It is crucial to get an accurate diagnosis because taking unnecessary iron supplements will not cure this type of anemia and can actually cause harmful iron buildup in the body. Treatment is rarely needed for this stage.
Hemoglobin H Disease
Missing three genes results in a clinical condition that requires active medical management. Patients with HbH disease need regular monitoring by a hematologist. During times of stress, illness, or pregnancy, their anemia can worsen suddenly, occasionally requiring blood transfusions to stabilize their health.
Hydrops Fetalis
When all four genes are missing, the situation is a medical emergency from the moment of conception. Modern medicine has made some strides in treating this through intrauterine blood transfusions (transfusions given to the fetus while still in the womb). However, these cases remain incredibly complex, requiring intense prenatal diagnosis and extremely specialized care immediately after birth.
Diagnosis of Alpha Thalassemia
Accurate diagnosis is the foundation of effective medical care. If a doctor suspects a blood disorder, they will use a combination of tests to pinpoint the exact issue.
Complete Blood Count (CBC) and Red Blood Cell Indices
The first step is usually a standard Complete Blood Count (CBC). This test measures the amount of hemoglobin in the blood and evaluates the size and shape of the red blood cells. A low hemoglobin count paired with unusually small red blood cells often points toward a thalassemia trait.
Hemoglobin Electrophoresis and HPLC
To get a clearer picture, doctors use tests like hemoglobin electrophoresis or High-Performance Liquid Chromatography (HPLC). These laboratory methods separate the different types of hemoglobin in the blood. By measuring the exact ratios of normal and abnormal proteins, doctors can confidently rule out other types of anemia. For a deeper dive into how these tests work, you can review this complete guide to diagnosing thalassemia.
Genetic Testing and DNA Analysis
The most definitive way to confirm the condition is through DNA testing. A genetic test can map out chromosome 16 and identify exactly how many alpha-globin genes are missing or mutated. This confirms the specific type of alpha thalassemia and provides essential information for future family planning.
Prenatal Diagnosis (Amniocentesis, Chorionic Villus Sampling)
For parents who know they are carriers, prenatal testing can determine if the fetus has inherited the disorder. Doctors can collect a small sample of the amniotic fluid or the placenta early in the pregnancy to analyze the baby’s DNA.
Management and Treatment Options
 Treatment plans are highly individualized. A person with a silent trait requires no intervention, while someone with HbH disease needs a structured medical strategy.
Treatment plans are highly individualized. A person with a silent trait requires no intervention, while someone with HbH disease needs a structured medical strategy.
Blood Transfusions (for severe forms)
For patients dealing with severe anemia, regular blood transfusions are a primary treatment. Transfusions provide the body with healthy red blood cells, instantly improving oxygen delivery, boosting energy levels, and suppressing the overactive bone marrow.
Iron Chelation Therapy (to manage iron overload)
Frequent blood transfusions introduce a new problem: excess iron. The body cannot naturally excrete the large amounts of iron found in transfused blood. Over time, this iron builds up and becomes toxic. To prevent organ failure, patients must take medications known as iron chelators. These drugs bind to the extra iron so the body can flush it out through urine. Recognizing liver iron overload symptoms early is critical for adjusting these medications properly.
Splenectomy (surgical removal of the spleen)
If the spleen becomes massively enlarged and starts destroying healthy red blood cells too quickly, a surgeon may need to remove it. A splenectomy can help stabilize hemoglobin levels, but it also increases the patient’s risk of severe infections, requiring them to stay up-to-date on all specific vaccinations.
Bone Marrow Transplantation (potential cure for some)
Currently, a bone marrow transplant (also called a stem cell transplant) is the only established cure for severe thalassemia. It involves replacing the patient’s defective blood-forming stem cells with healthy ones from a matching donor. This is a high-risk procedure usually reserved for the most severe cases and requires a perfectly matched sibling donor for the best results.
Folic Acid Supplementation
Folic acid is a B vitamin that helps build healthy red blood cells. Because patients with HbH disease have bone marrow that works constantly to replace destroyed cells, their bodies burn through folate quickly. A daily folic acid supplement is an easy and effective way to support red blood cell production.
Lifestyle Management and Supportive Care
Medical treatments work best when paired with healthy daily habits. Patients are encouraged to avoid taking any over-the-counter vitamins containing iron unless explicitly told to do so by their hematologist.
Living with Alpha Thalassemia
A chronic illness diagnosis requires lifestyle adjustments, but with the right support system, patients can lead fulfilling, active lives.
Nutritional considerations and dietary advice
Diet plays a supportive role in overall health. Patients, especially those receiving transfusions, should generally avoid iron-heavy foods like fortified cereals or massive portions of red meat. Drinking tea with meals can also naturally decrease the amount of iron the body absorbs from food.
Importance of regular medical check-ups and monitoring
Consistency is key. Patients with symptomatic alpha thalassemia must attend regular appointments to monitor their hemoglobin levels, check their liver and heart function, and evaluate their bone health. Routine monitoring allows doctors to adjust treatment plans before small issues turn into major complications. Resources from the Centers for Disease Control and Prevention (CDC) provide excellent guidelines on what these regular check-ups should include.
Psychological and emotional support
Managing a lifelong condition can take a toll on mental health. Anxiety, fatigue, and the stress of frequent medical appointments are heavy burdens. Connecting with mental health professionals or joining patient support groups provides a safe space to share frustrations and learn coping strategies.
Genetic counseling and family planning
Because this is an inherited condition, genetic counseling is highly recommended for anyone with the trait who is considering starting a family. A counselor can explain the exact probabilities of passing the disorder to a child and discuss all available testing and family planning options. If you want to learn more about the broader implications of these genetic traits, you can explore resources focused on understanding alpha thalassemia symptoms.
Early diagnosis and prevention strategies
Public health initiatives are focusing heavily on early screening. By identifying carriers before they have children, healthcare systems can drastically reduce the number of severe hydrops fetalis cases. Organizations like the National Heart, Lung, and Blood Institute (NHLBI) continue to fund research that makes these diagnostic tools faster, cheaper, and more accessible worldwide.








{kind=link}