Alpha thalassemia symptoms vary based on gene changes and can range from no signs to severe anemia. Common effects include tiredness, pale skin, weakness, and enlarged spleen. Early detection helps in better management and health monitoring.
Understanding Alpha Thalassemia: The Genetic Basis
To understand why certain alpha thalassemia symptoms occur, it is important to first look at how hemoglobin is formed in the body. Hemoglobin, the protein responsible for carrying oxygen in red blood cells, is made up of two essential components: alpha-globin and beta-globin chains. A healthy balance between these two is necessary for proper oxygen transport and normal body function.
The Role of Alpha-Globin Genes
The human body depends on four alpha-globin genes to produce the alpha protein chains needed for hemoglobin. Two of these genes are inherited from each parent. In alpha thalassemia, the severity of the condition depends on how many of these four genes are missing or altered, which is part of the broader types of thalassemia.
When all four genes are functioning properly, hemoglobin production remains normal. However, when one or more genes are affected, the production of alpha chains decreases. This imbalance disrupts normal blood function and leads to different forms of alpha thalassemia within the overall types of thalassemia spectrum, ranging from silent carriers to more severe conditions.
How Gene Changes Lead to the Condition
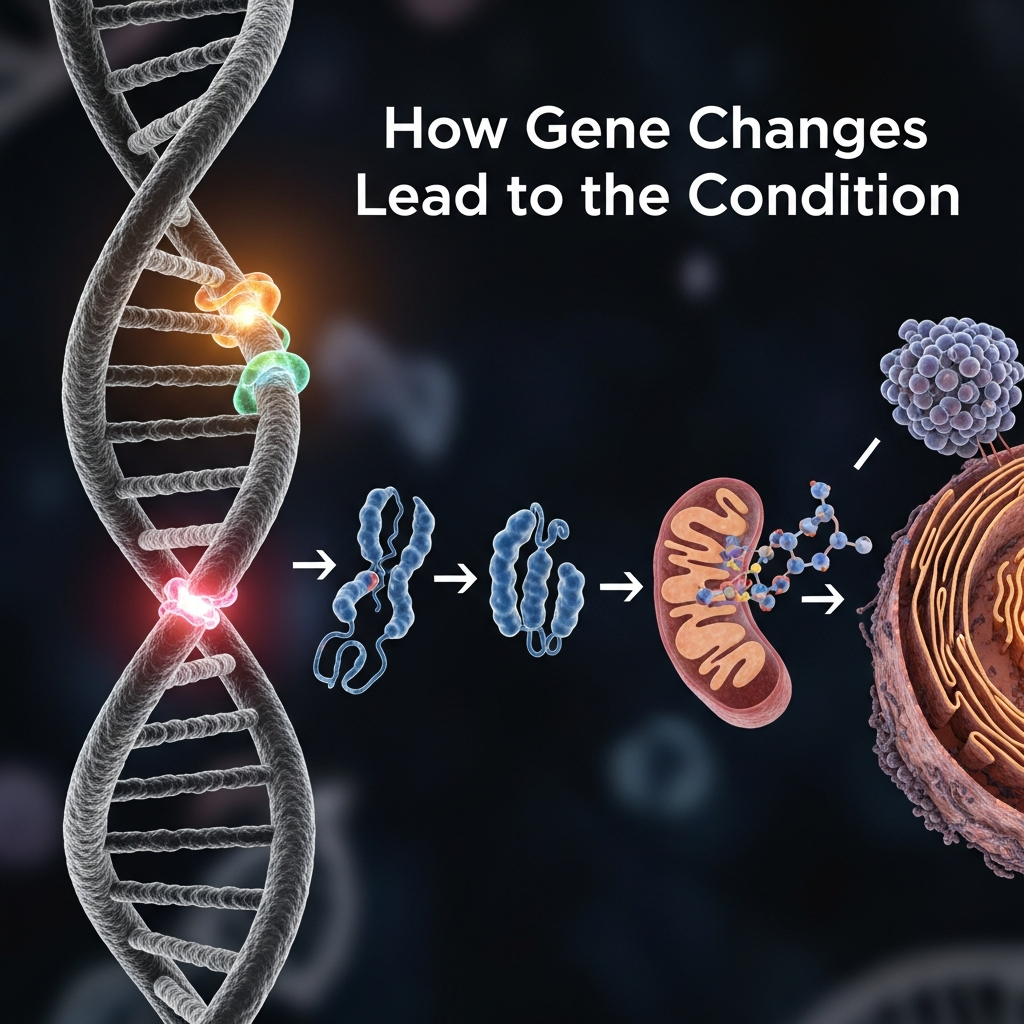 When alpha-globin genes are deleted or damaged, the body is unable to produce enough alpha protein chains. As a result, excess beta chains begin to accumulate and form unstable clusters. These abnormal structures damage red blood cells, causing them to break down more quickly than normal.
When alpha-globin genes are deleted or damaged, the body is unable to produce enough alpha protein chains. As a result, excess beta chains begin to accumulate and form unstable clusters. These abnormal structures damage red blood cells, causing them to break down more quickly than normal.
This rapid destruction of red blood cells leads to a shortage of healthy oxygen-carrying cells in the body. As a result, individuals develop common alpha thalassemia symptoms such as tiredness, pale skin, weakness, and in more serious cases, organ enlargement or growth problems. In some individuals, alpha thalassemia symptoms may be very mild or even unnoticed, while in others they can significantly impact daily life. Recognizing these alpha thalassemia symptoms early can help in seeking timely medical attention and preventing complications. The severity of these symptoms depends on how many genes are affected and the specific type of thalassemia present.
Importance of Understanding the Genetic Process
Understanding this genetic process is essential for recognizing early warning signs, diagnosing the condition correctly, and managing different types of thalassemia effectively. Awareness of alpha thalassemia symptoms also plays a key role in early detection and proper treatment planning.
Early awareness helps in preventing severe complications through timely medical care, proper screening, and genetic counseling for families at risk. By understanding both the genetic causes and the visible alpha thalassemia symptoms, individuals and healthcare providers can work together to improve long-term health outcomes.
The Spectrum of Alpha Thalassemia Symptoms
Because the condition depends on the exact number of affected genes, patients experience a massive variance in physical signs. Medical professionals categorize the disease into four main tiers based on genetic deletion.
Silent Carrier State (Alpha Thalassemia Minima)
When only one of the four alpha-globin genes is missing, the individual is considered a silent carrier.
- Number of affected genes: One.
- Lack of noticeable symptoms: Silent carriers produce almost normal amounts of hemoglobin. As the name suggests, they generally experience zero health issues and have no outward alpha thalassemia symptomps.
- Diagnosis and genetic counseling: Blood tests usually appear completely normal. The condition is often only discovered through specialized genetic DNA testing, which is highly recommended before starting a family to understand the odds of passing the missing gene to children.
Alpha Thalassemia Trait (Alpha Thalassemia Minor)
When two genes are missing, the diagnosis is alpha thalassemia trait. The two missing genes can be inherited from the same parent or one from each parent.
- Number of affected genes: Two.
- Mild anemia symptoms: Patients typically experience mild, manageable signs of anemia, such as occasional fatigue and slight pallor.
- Differentiating from iron deficiency anemia: Because the red blood cells appear small under a microscope, doctors frequently misdiagnose this trait as iron deficiency. Iron supplements will not cure thalassemia trait and can actually be harmful if overused.
- Importance of genetic testing: Accurately identifying this trait through genetic testing helps clarify the specific types of thalassemia present in a family tree, guiding better prenatal care.
Hemoglobin H Disease
A much more serious condition arises when three genes are deleted or severely mutated. This leads to an abundance of beta-globin proteins that form abnormal structures called Hemoglobin H.
- Number of affected genes: Three.
- Moderate to severe anemia symptoms: Patients experience a pronounced lack of oxygen. Common alpha thalassemia symptomps at this stage include chronic fatigue, jaundice (yellowing of the skin and eyes), enlarged spleen (splenomegaly), and pronounced bone changes in the face due to the bone marrow working overtime.
- Complications: The rapid breakdown of red blood cells can lead to gallstones. Furthermore, the body naturally absorbs more iron from food to try and fix the anemia, which often results in dangerous iron overload.
- Management and treatment options: Patients require close medical supervision, occasional blood transfusions, and careful monitoring of organ function.
Hydrops Fetalis (Alpha Thalassemia Major)
The most severe form of the disorder occurs when all four alpha-globin genes are missing.
- Number of affected genes: Four.
- Life-threatening condition: Without alpha-globin, normal hemoglobin simply cannot form. This condition is overwhelmingly fatal to the fetus before or shortly after birth, and it poses severe health risks to the pregnant mother, including high blood pressure and dangerous bleeding.
- Symptoms in utero: The fetus develops severe anemia, leading to massive fluid accumulation throughout the body and eventual heart failure.
- Diagnosis and prenatal interventions: Ultrasound monitoring and early prenatal DNA testing are critical. In highly rare, carefully managed cases, intrauterine blood transfusions have allowed babies to survive until birth, though they require lifelong intensive medical care.
Diagnosing Alpha Thalassemia
 Early and accurate diagnosis plays a key role in identifying this blood disorder and preventing complications later in life. Since symptoms can overlap with other forms of anemia, doctors use a combination of routine screening and advanced laboratory tests to confirm the condition and understand its severity within the broader types of thalassemia.
Early and accurate diagnosis plays a key role in identifying this blood disorder and preventing complications later in life. Since symptoms can overlap with other forms of anemia, doctors use a combination of routine screening and advanced laboratory tests to confirm the condition and understand its severity within the broader types of thalassemia.
Complete Blood Count (CBC)
A Complete Blood Count is usually the first step in evaluation. It measures hemoglobin levels and examines the size and shape of red blood cells. In many cases, smaller and paler red blood cells raise suspicion of a possible inherited blood disorder. While this test alone cannot confirm alpha thalassemia, it provides an important early indication.
Hemoglobin Electrophoresis
Hemoglobin electrophoresis, often combined with High-Performance Liquid Chromatography (HPLC), is used to analyze different forms of hemoglobin in the blood. This helps doctors distinguish between alpha thalassemia and beta thalassemia and other related blood conditions. It provides valuable information about abnormal hemoglobin patterns and overall oxygen-carrying capacity.
Genetic Testing for Gene Deletions
Genetic testing is the most accurate method for confirming diagnosis. By examining DNA, doctors can identify exactly how many alpha-globin genes are missing or altered. This test is essential for understanding disease severity and for classifying different forms within the types of thalassemia spectrum.
Family History and Genetic Counseling
A detailed family history is extremely important in diagnosing inherited blood disorders. Since alpha thalassemia is passed from parents to children, genetic counseling helps families understand the risk of transmission. It also plays a vital role in guiding reproductive decisions and early screening for future generations.
Management and Treatment Approaches
Treatment depends on how severe the condition is. Some individuals may remain healthy carriers without needing treatment, while others with more serious forms require long-term medical care. Management strategies vary across both alpha thalassemia and beta thalassemia cases.
Blood Transfusions
In moderate to severe cases, regular blood transfusions are used to provide healthy red blood cells. This helps reduce symptoms like fatigue, weakness, and poor growth. It also supports normal development in children and improves overall quality of life.
Iron Chelation Therapy
Repeated transfusions and increased iron absorption can lead to excess iron buildup in vital organs. Iron chelation therapy helps remove this excess iron from the body using special medications. Proper iron control is essential to prevent long-term damage to the heart, liver, and endocrine system.
Folic Acid Supplementation
Folic acid supports red blood cell production and helps the bone marrow function more effectively. Doctors often recommend it as a supportive treatment to improve blood health and reduce the strain on the body.
Splenectomy
In some severe cases, the spleen becomes overactive and enlarges as it tries to remove damaged red blood cells. This can worsen anemia over time. In carefully selected patients, removing the spleen may help improve red blood cell survival and reduce complications.
Bone Marrow or Stem Cell Transplant
A bone marrow transplant is currently the only potential curative option for severe cases. It replaces defective blood-forming cells with healthy ones from a matched donor. After successful transplantation, the body may begin producing normal hemoglobin, reducing or eliminating symptoms.
Prenatal Diagnosis
For families with a known history of inherited blood disorders, prenatal testing is available. Procedures such as chorionic villus sampling or amniocentesis can detect whether a fetus has inherited gene changes. This helps families make informed medical and personal decisions early in pregnancy.
Living with Alpha Thalassemia: Challenges and Support
Living with a chronic inherited blood condition involves more than just medical treatment. It affects daily routines, emotional well-being, and social life for both patients and their families. Understanding these challenges is an important part of managing long-term care in different types of thalassemia, including alpha thalassemia and beta thalassemia.
Impact on Quality of Life
Regular hospital visits for monitoring, blood transfusions, and laboratory tests can significantly affect daily life. These ongoing medical needs may interrupt education, employment, and social activities, especially in children and young adults.
In addition, persistent tiredness caused by reduced oxygen delivery in the body can limit physical activity. Many individuals need to carefully balance rest and activity to maintain their energy levels and overall health, especially in more severe forms of thalassemia.
Psychological and Emotional Support
Living with a lifelong genetic condition can sometimes lead to emotional stress, anxiety, or feelings of uncertainty about the future. This impact is often seen in both patients and their caregivers.
Access to counseling services, mental health professionals, and strong family support systems plays an important role in coping with the condition. Emotional care is just as important as physical treatment in improving overall well-being for people affected by different types of thalassemia.
The Role of Patient Organizations
Support groups and patient organizations provide valuable assistance to individuals and families. These groups offer education about the condition, help patients connect with specialized healthcare providers, and create a sense of community.
Sharing experiences with others facing similar challenges can reduce feelings of isolation and provide practical advice for daily life management. These networks are especially helpful for families dealing with alpha thalassemia and beta thalassemia.
Research and Future Directions
Medical research continues to improve understanding and treatment options for inherited blood disorders. Scientists are working on more advanced and less invasive approaches that may improve long-term outcomes for people with different types of thalassemia.
Gene-Based Treatments
Gene therapy is one of the most promising areas of research. Scientists are exploring methods to correct or replace faulty genetic instructions in blood-forming cells. If successful, this approach could offer long-term improvement rather than lifelong symptom management.
New Medical Treatments
Researchers are also developing new medications that aim to improve red blood cell function or stimulate alternative forms of hemoglobin production. These treatments may help reduce symptoms and decrease the need for frequent medical procedures in both alpha thalassemia and beta thalassemia cases.
Advances in Early Detection
Improved genetic screening technologies are making it easier and faster to identify inherited blood conditions. Early detection allows for better planning, timely treatment, and informed family decisions before symptoms become severe across different types of thalassemia.
Taking Control of Your Genetic Health
 Alpha thalassemia is a complex inherited blood condition that requires awareness, early evaluation, and ongoing care. Because alpha thalassemia symptoms can range from completely unnoticed to severe and life-threatening, understanding your genetic background becomes an important step in protecting long-term health.
Alpha thalassemia is a complex inherited blood condition that requires awareness, early evaluation, and ongoing care. Because alpha thalassemia symptoms can range from completely unnoticed to severe and life-threatening, understanding your genetic background becomes an important step in protecting long-term health.
In many cases, people may not realize they carry a gene change until routine blood tests or family screening reveal it. This is why learning about inherited blood conditions and the different types of thalassemia is important, especially since severity and symptoms like alpha thalassemia symptoms can vary widely from person to person.
If you have a family history of unexplained anemia, frequent fatigue, or known blood-related conditions, it is highly recommended to seek medical advice. Planning a family is another important stage where genetic awareness plays a key role. A consultation with a genetic counselor or a blood specialist can help identify risks early and guide appropriate testing for both alpha thalassemia and beta thalassemia.
Through proactive screening, accurate diagnosis, and personalized care plans, individuals can better manage their health and avoid complications. With proper medical support and regular monitoring, people affected by this condition can still lead stable, productive, and fulfilling lives while staying informed about their genetic health.
Frequently Asked Questions
1. What is alpha thalassemia?
It is one of the types of thalassemia, an inherited blood disorder that affects hemoglobin production and causes varying levels of anemia.
2. What causes alpha thalassemia?
It is caused by changes or missing genes responsible for alpha globin production, which leads to reduced oxygen-carrying capacity in the blood.
3. What are the common alpha thalassemia symptoms?
Symptoms can range from none in mild cases to fatigue, pale skin, weakness, and in severe cases, organ enlargement and growth problems.
4. How is it different from other types of thalassemia?
It differs from beta thalassemia based on which globin chain is affected and how many genes are involved, leading to different severity levels.
5. Can someone have it without symptoms?
Yes, some individuals are silent carriers and may not show noticeable signs but can still pass the condition to their children.
6. How is alpha thalassemia diagnosed?
It is diagnosed using blood tests like CBC, hemoglobin analysis, and genetic testing to confirm gene changes.
7. Is alpha thalassemia inherited?
Yes, it is passed from parents to children through genetic inheritance patterns.
8. Can it be cured?
In some severe cases, a stem cell or bone marrow transplant may offer a potential cure, but most treatment focuses on management.
9. Does it require lifelong treatment?
Mild forms may not need treatment, but moderate to severe types of thalassemia often require long-term medical care.
10. Can people live a normal life with it?
Yes, with early diagnosis, proper care, and regular monitoring, many individuals can live healthy and productive lives.
Conclusion
Alpha thalassemia is a genetic condition that belongs to the broader types of thalassemia, ranging from mild carrier states to more serious health forms. Its impact depends on how many genes are affected and how the body produces hemoglobin.
While some people may experience only mild or no alpha thalassemia symptoms, others may require ongoing medical support depending on severity. Advances in modern healthcare have significantly improved management options, making it possible for many patients to live stable lives.
Most importantly, awareness of the types of thalassemia, early screening, and genetic counseling play a crucial role in prevention and early intervention. With proper medical guidance and care, individuals affected by this condition can lead healthy, meaningful lives.










{kind=link}